Solitary fibrous tumor of the pleura: clinical case report
Article / Artículo
https://doi.org/10.33821/793
Date received: 20/2/2024
Date accepted: 10/6/2024
1. Introduction
Solitary fibrous tumors (SFT) are a rare neoplasm of mesenchymal origin, accounting
for less than 2% of all soft tissue tumors, with an estimated annual incidence of
one case per million people. It occurs predominantly in adults between the fifth and
sixth decades of life, with no gender preference (1). The most common locations are deep somatic soft tissues and body cavities (especially
the pleura and abdominal cavity) (2). The World Health Organization's Classification of Tumors of Soft Tissue and Bone,
fifth edition (WHO, 2020), categorizes it within tumors of fibroblastic origin, avoiding
the terms "typical" or "malignant," given that even cases with benign-appearing histology
can progress. The risk of recurrence or metastasis has been reported to range from
5% to 10%, typically to the lungs, liver, and bone (2). The estimated 5- and 10-year overall survival rates are 89% and 73%, respectively,
and the 5- and 10-year recurrence-free survival rates are 74% and 55%, highlighting
the importance of long-term surveillance (2,3).
Through the presentation of a clinical case of a large pleural tumor, the application
of histopathological and molecular criteria that confirm the diagnosis is addressed,
and the inclusion of the mDemicco scale for metastasis risk stratification reinforces
the importance of prolonged surveillance. In essence, this case provides evidence
of the usefulness of these diagnostic and follow-up tools, which is of great value
to the medical community, given the rarity of the disease.
2. Clinical case
A 53-year-old female patient presented with a history of hypothyroidism treated with
levothyroxine 100 mcg daily and stage IA2 cervical cancer treated with radical hysterectomy
in 2005. There was no known family history of cancer. Denied smoking or exposure to
environmental toxins. She presented to the Pneumology Department in May 2019 with
a nonproductive cough lasting more than three months and progressive dyspnea. Initial
physical examination revealed a respiratory rate of 22 rpm, oxygen saturation of 95%
on room air, hypoventilation in the left lung base on pulmonary auscultation, and
dullness to percussion in the left basal region. Tumor marker results were as follows:
CYFRA 21-1: 1.85 ng/ml; NSE: 19.51 ng/ml. Pulmonary function tests evidenced a moderate
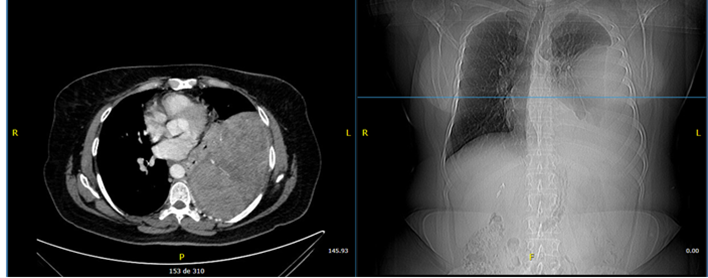
restrictive pattern with no response to bronchodilators. Contrast-enhanced chest CT
scan showed a heterogeneous extrapulmonary mass measuring 12.8 x 15.3 cm with a broad
base on the left parietal pleura and no adenomegaly (Figure 1).
Figure 1
Contrast-enhanced chest CT scan. Left extrapulmonary heterogeneous lesion with an
obtuse angle, which measures 12.8 x 15.3 cm.
A CT-guided percutaneous biopsy was performed, which showed the presence of spindle
cells arranged in a storiform pattern. This histological pattern is classic in soft
tissue pathology and points the differential diagnosis mainly toward tumors such as
solitary fibrous tumor, dermatofibrosarcoma protuberans, and undifferentiated pleomorphic
sarcoma. Immunohistochemical analysis showed positivity for BCL-2 and CD34 markers.
These results, together with the clinical-imaging correlation, confirmed the diagnosis
of solitary fibrous tumor. Consequently, the proposed therapeutic approach was surgical
resection. A left thoracotomy was performed with complete resection of a 23 cm lobulated
tumor originating in the parietal pleura without pulmonary or bone invasion.
The pathological report revealed spindle cells arranged in a storiform pattern with
minimal nuclear atypia and moderate mitotic activity (5/10 fields), which is a common
pattern in SFT, as well as positivity for CD34 and BCL2 immunophenotypes of SFT. Although
the absence of the STAT6 marker is a limitation of the study, the combination of the
other histological findings and positivity for these markers, in the context of a
pleural tumor, is highly suggestive. It is important to note that dermatofibrosarcoma
protuberans (DFSP) was ruled out because, in addition to being a neoplasm that usually
originates in the dermis and subcutaneous tissue, it presents an infiltrative honeycomb
pattern and low mitotic activity, characteristics that were not observed in this case.
Furthermore, DFSP usually expresses CD34 but lacks the histological pattern characteristic
of SFT ("star" or "made of rag" pattern, hemangiopericytoma-like branching vessels,
and areas of hyalinized collagen), which allowed it to be excluded in correlation
with the pleural location of the tumor.
Pleomorphic undifferentiated sarcoma (formerly known as malignant fibrous histiocytoma)
was ruled out based on the immunohistochemistry result, as this type of sarcoma typically
does not express CD34 or BCL2, whereas in the present case, both proteins were positive,
a finding characteristic of solitary fibrous tumor. Additionally, pleomorphic sarcoma
usually shows marked cellular pleomorphism, abundant atypia, and a high mitotic index;
features that were absent in the patient's histological evaluation.
The pleural location, the exophytic size of 23 cm, and the resected block specimen
without macroscopic infiltration favor a pleural SFT, consistent with the histological
and immunohistochemical findings already described.
Figure 2
Macroscopic findings. Lobulated tumor measuring 22,9 X 15,6 X 9 cm.
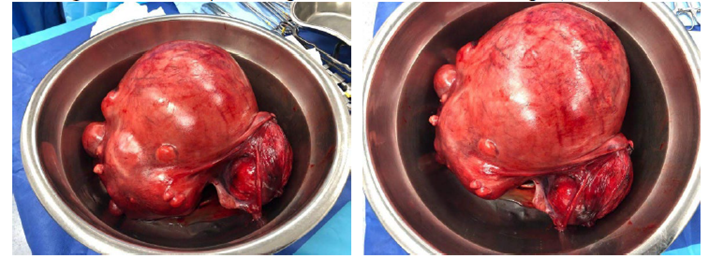
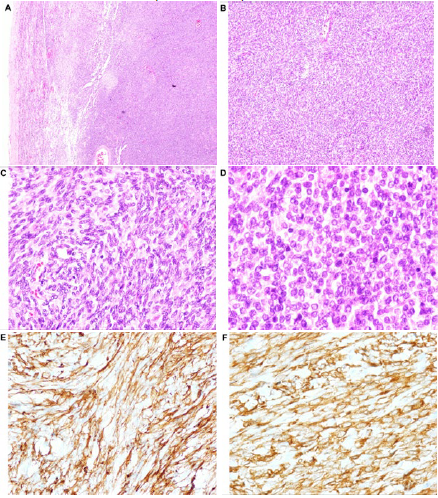
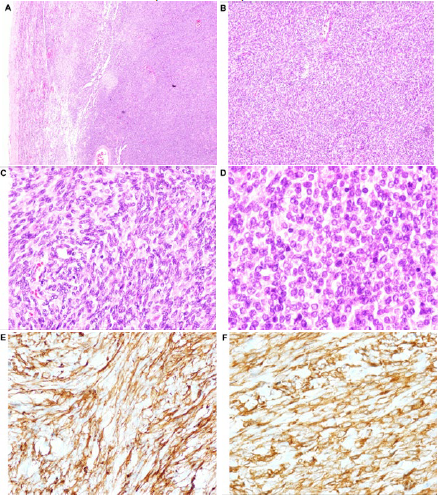
Figure 3
Histological findings. A. Highly fusocellular vascularized, solid neoplasm (H&E, 5x magnification). B. Cells distributed in short fascicles with a storiform pattern; no areas of necrosis
were identified (H&E, 10x magnification). C. Vesicular nuclei with inconspicuous nucleoli and mild nuclear atypia (H&E, 20x magnification).
D. Atypical mitoses, 5/10 fields at 40x (H&E, 60x magnification). E. CD34 positive. F. BCL2 positive.
Risk stratification for recurrence using the mDemicco scale yielded a score of 5,
classifying the patient as intermediate risk. Adjuvant treatment was not indicated
given the negative surgical margins and intermediate risk stratification. The patient
remains under clinical follow-up with serial chest CT scans. At six years post-operatively,
the patient remained asymptomatic with stable respiratory function tests and no evidence
of recurrence. The patient demonstrated good adherence to follow-up and no adverse
events related to surgical treatment were observed.
3. Discussion
Pleural solitary fibrous tumors (SFT) are rare, usually slow-growing mesenchymal neoplasms
that can reach large dimensions before producing clinical symptoms, which are generally
compressive (4). The etiology remains unknown, and no association with environmental factors such
as tobacco use or asbestos exposure has been demonstrated (3). Diagnosis requires imaging studies combined with histopathological and immunohistochemical
analysis. Complete surgical resection with negative margins is the treatment of choice,
and there is a better prognosis when complete tumor resection with clear surgical
margins is achieved. However, some cases may have an unpredictable course; therefore,
long-term follow-up is mandatory (5).
Classic histopathological findings include randomly arranged spindle-shaped to ovoid
cells, with branching blood vessels, thin deer antler-shaped walls, and prominent
stromal collagen, taking into account the mitotic count and the percentage of tumor
necrosis (2). Nuclear expression of STAT6 has been shown to be highly specific for SFT, which
is a direct consequence of the NAB2-STAT6 gene fusion and is detectable by immunohistochemistry.
In retrospective studies evaluating localized disease, the fusion between exon 4 of
NAB2 and exon 2 of STAT6 was the most frequent variant, which has been correlated
with a thoracic location, older patient age, lower mitotic count, and better prognosis.
On the other hand, the fusion between exon 6 of NAB2 and exon 16 of STAT6, identified
as the second most common fusion, is associated with extrathoracic locations, young
patients, and a worse prognosis (6).
In some studies, CD209 overexpression has shown a significant correlation with an
unfavorable clinical prognosis (6). Additionally, evaluation of the Ki-67 proliferation index is useful for predicting
prognosis (7). Other markers, although nonspecific, such as CD34, BCL-2, and CD99 expression,
may also be present; however, CD34 may be negative in approximately 10%, while CD99
and BCL2 are often expressed in other neoplasms (8).
To predict the risk of recurrence, the mDemicco scale is used, based on four parameters
such as age (>55 years: 1 point), tumor size (5-10 cm: 1 point, 10-15 cm: 2 points,
and ≥ 15 cm: 3 points), mitotic index (1 to 3 mitoses per 10 fields: 1 point, and
≥ 4 mitoses per 10 fields: 2 points), and percentage of tumor necrosis (≥ 10%: 1 point).
Based on this score, the risk is classified as low (0-3 points), intermediate (4-5
points), or high (6-7 points) (7,9). Complete surgical resection is the cornerstone of treatment for SFT. However, postoperative
management should be tailored individually, considering surgical margins and risk
stratification score. Although data are limited, neoadjuvant chemotherapy and radiotherapy
may be considered in advanced cases. In cases of intermediate to high risk with positive
margins and unresectable/inoperable tumors, adjuvant RT is a reasonable option (10). Likewise, the use of the tyrosine kinase inhibitor (TKI) pazopanib could be considered
as first-line systemic treatment in these cases (6). Without a doubt, all treatment decisions should be made by a multidisciplinary
team.
4. Conclusion
This report highlights that early diagnosis, complete resection, and a structured
follow-up plan are crucial for achieving favorable clinical outcomes in solitary fibrous
tumors of the pleura. Although the tumor was large, the postoperative course was uneventful,
and long-term follow-up showed no evidence of recurrence. These findings underscore
the key role of a multidisciplinary approach and close monitoring in the management
of this entity.
1. Introducción
El tumor fibroso solitario (TFS) es una neoplasia poco frecuente de origen mesenquimatoso,
que representa menos del 2 % de todos los tumores de tejidos blandos, con una incidencia
anual estimada de un caso por millón de personas. Se presenta predominantemente en
adultos entre la quinta y la sexta década de la vida, sin preferencia de género [1]. Las localizaciones más frecuentes son los tejidos blandos somáticos profundos y
las cavidades corporales, sobre todo la pleura y la cavidad abdominal [2]. La Clasificación de Tumores de Tejidos Blandos y Huesos de la Organización Mundial
de la Salud quinta edición [3] lo categoriza dentro de los tumores de origen fibroblástico y evita los términos
típico o maligno, dado que incluso los casos con histología de apariencia benigna pueden progresar.
Se ha reportado que el riesgo de recidiva o metástasis oscila entre el 5 y el 10 %,
típicamente a pulmones, hígado y hueso [2]. Se estima que las tasas de supervivencia global a 5 y 10 años son del 89 y del
73 %, respectivamente, y la supervivencia libre de recurrencia a 5 y 10 años es del
74 y el 55 %, lo que resalta la importancia de una vigilancia a largo plazo [2,4].
A través de la presentación de un caso clínico de un tumor pleural de gran tamaño,
se aborda la aplicación de criterios histopatológicos y moleculares que confirman
el diagnóstico. La inclusión de la escala mDemicco para la estratificación del riesgo
de metástasis refuerza la importancia de la vigilancia prolongada. En esencia, este
caso aporta evidencia sobre la utilidad de estas herramientas de diagnóstico y seguimiento,
lo que es de gran valor para la comunidad médica, dada la rareza de la enfermedad.
2. Caso clínico
Paciente femenina de 53 años con antecedentes de hipotiroidismo en tratamiento con
levotiroxina 100 mcg/día y cáncer de cuello uterino estadio IA2 tratado mediante histerectomía
radical en el 2005. Sin antecedentes familiares oncológicos conocidos. Niega hábito
tabáquico o exposición a tóxicos ambientales. Consulta al departamento de Neumología
en mayo del 2019 por tos no productiva de más de tres meses de evolución y disnea
progresiva. En el examen físico inicial presentó frecuencia respiratoria de 22 rpm,
saturación de oxígeno 95 % a aire ambiente; en la auscultación pulmonar, hipoventilación
en base pulmonar izquierda y percusión mate en región basal izquierda. Resultados
de marcadores tumorales: Fragmentos de citoqueratina 19 (CYFRA 21-1): 1,85 ng/ml;
Enolasa Neuronal Específica (NSE): 19,51 ng/ml. Las pruebas de función pulmonar reportaron
patrón restrictivo moderado sin respuesta a broncodilatador. La tomografía torácica
con contraste reportó una masa extrapulmonar heterogénea, de 12,8 × 15,3 cm, con base
amplia en pleura parietal izquierda, no adenomegalias (Figura 1).
Figura 1
Tomografía contrastada de tórax. Lesión heterogénea extrapulmonar izquierda de ángulo
obtuso que mide 12,8 × 15,3 cm.
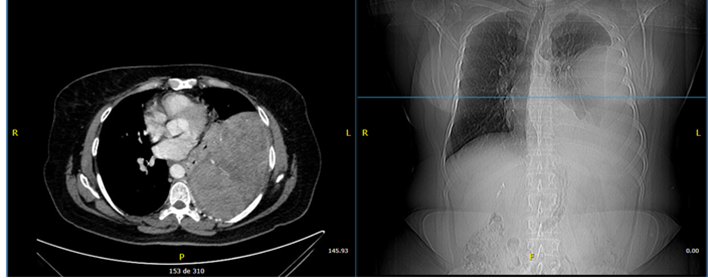
Se practicó una biopsia percutánea guiada por tomografía que reportó la presencia
de células fusiformes dispuestas en un patrón estoriforme. Este patrón histológico
es clásico en la patología de tejidos blandos y orienta el diagnóstico diferencial
principalmente hacia tumores como el fibroso solitario, el dermatofibrosarcoma protuberans
(DFSP) y el sarcoma pleomórfico indiferenciado. El análisis de inmunohistoquímica
demostró positividad para los marcadores BCL-2 y CD34. Estos resultados, en conjunto
con la correlación clínico-imagenológica, consolidaron el diagnóstico de tumor fibroso
solitario. En consecuencia, el abordaje terapéutico propuesto fue la resección quirúrgica.
Se realizó una toracotomía izquierda con resección completa de tumor lobulado de 23
cm, originado en la pleura parietal costal sin invasión pulmonar ni ósea. El informe
anatomopatológico reportó la presencia de células fusiformes en un patrón estoriforme
con escasa atipia nuclear y actividad mitótica moderada (5/10 campos), un patrón común
en el TFS, además de la positividad para CD34 y BCL2, inmunofenotipos del TFS. Aunque
la ausencia del marcador STAT6 es una limitación del estudio, la combinación del resto
de los hallazgos histológicos y la positividad para estos marcadores, en un contexto
de un tumor pleural, es altamente sugestivo. Es importante señalar que se descartó
el DFSP debido a que, además de ser una neoplasia que habitualmente se origina en
la dermis y el tejido subcutáneo, presenta un patrón infiltrativo en panal de abeja
y escasa actividad mitótica, características que no se observaron en este caso. Asimismo,
el DFSP suele expresar CD34, pero carece del patrón histológico característico del
TFS (patrón en “estrellas” o “hecho de trapo”, vasos ramificados tipo hemangiopericitoma
y áreas de colágeno hialinizado), lo cual permitió excluirlo en correlación con la
localización pleural del tumor.
Por otra parte, el sarcoma pleomórfico indiferenciado, antes denominado histiocitoma
fibroso maligno, fue descartado con el resultado de la inmunohistoquímica, ya que
este tipo de sarcoma típicamente no expresa CD34 ni BCL2, mientras que en el presente
caso ambas proteínas fueron positivas, hallazgo característico del TFS. Adicionalmente,
el sarcoma pleomórfico suele mostrar marcado pleomorfismo celular, abundante atipia
y alto índice mitótico, rasgos ausentes en la evaluación histológica del paciente.
La localización pleural, el tamaño exofítico de 23 cm y la pieza resecada en bloque
sin infiltración macroscópica favorecen un TFS pleural, en coherencia con los hallazgos
histológicos e inmunohistoquímicos ya descritos.
Figura 2
Hallazgos macroscópicos. Tumoración lobulada de 22,9 × 15,6 × 9 cm.
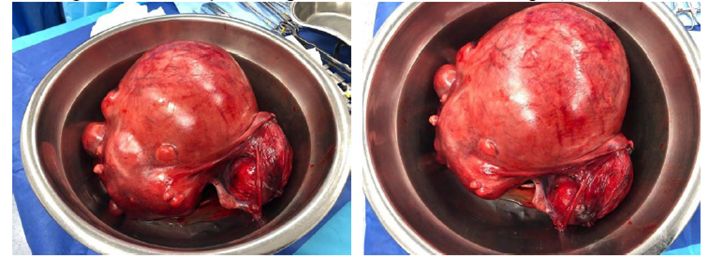
Figura 3
Hallazgos histológicos. A. Neoplasia altamente fusocelular vascularizada, sólida (H-E, aumento 5x). B. Células distribuidas en fascículos cortos y con patrón estoriforme, no se identificaron
áreas de necrosis (H-E, aumento 10x). C. Núcleos vesiculosos con nucleolo inconspicuo y atipia nuclear leve (H-E, aumento
20x). D. Mitosis atípicas, 5/10 campos de 40 x (H-E, aumento 60x). E. CD34 positivo. F. BCL2 positivo.
La estratificación de riesgo de recurrencia con la escala mDemicco resultó en un puntaje
de 5; la paciente fue clasificada en la categoría de riesgo intermedio. No se indicó
tratamiento adyuvante debido a márgenes negativos y estratificación de riesgo intermedio.
La paciente se mantiene en seguimiento clínico y es evaluada con tomografías de tórax
seriadas. Han transcurrido seis años desde la cirugía y permanece asintomática, con
pruebas de función respiratoria estables y sin evidencia de recidiva. Se confirmó
buena adherencia al seguimiento y ausencia de eventos adversos relacionados con el
tratamiento quirúrgico.
3. Discusión
El TFS pleural es una neoplasia mesenquimatosa infrecuente, generalmente de crecimiento
lento, que puede alcanzar grandes dimensiones antes de producir síntomas clínicos,
generalmente compresivos [5]. La etiología es desconocida, no se ha demostrado asociación con factores ambientales
como consumo de tabaco o exposición al asbesto [4]. El diagnóstico requiere estudios imagenológicos, hallazgos histopatológicos e inmunohistoquímicos.
La resección quirúrgica completa con márgenes negativos es el tratamiento de elección,
pues muestra un mejor pronóstico cuando se consigue una resección tumoral completa
con márgenes quirúrgicos libres, a pesar de ello algunos pueden tener un curso impredecible,
por lo que el seguimiento a largo plazo es obligatorio [6].
Los hallazgos histopatológicos clásicos son la presencia de células fusiformes a ovoides
dispuestas al azar, con vasos sanguíneos ramificados, de paredes delgadas en forma
de cuerno de ciervo y colágeno estromal prominente, tomando en cuenta el recuento
mitótico y el porcentaje de necrosis tumoral [2]. Se ha demostrado que la expresión nuclear de STAT6 es altamente específica para
el TFS, la cual es consecuencia directa de la fusión génica NAB2-STAT6 y es detectable
mediante inmunohistoquímica. En estudios retrospectivos que evaluaron la enfermedad
localizada, la fusión entre el exón 4 de NAB2 y el exón 2 de STAT6 fue la variante
más frecuente, la cual se ha correlacionado con una localización torácica, mayor edad
del paciente, un menor recuento mitótico y un mejor pronóstico. Por otro lado, la
fusión entre el exón 6 de NAB2 y el exón 16 de STAT6, identificada como la segunda
fusión más común, se ha asociado con localizaciones extratorácicas, pacientes jóvenes
y un peor pronóstico [7].
En algunos estudios, la sobreexpresión de CD209 ha mostrado una correlación significativa
con un pronóstico clínico desfavorable [7]. Adicionalmente, la evaluación del índice de proliferación Ki-67 es útil para predecir
el pronóstico [8]. Otros marcadores, aunque inespecíficos, como la expresión de CD34, BCL-2 y CD99,
también pueden estar presentes; sin embargo, CD34 puede ser negativo en aproximadamente
el 10 %, mientras que CD99 y BCL2 suelen expresarse en otras neoplasias [9].
Para predecir el riesgo de recurrencia, se emplea la escala mDemicco, basada en cuatro
parámetros: edad (> 55 años: 1 punto); tamaño tumoral (5-10cm: 1 punto, 10-15cm: 2
puntos y ≥ 15cm: 3 puntos); índice mitótico (1 a 3 mitosis por 10 campos: 1 punto
y ≥ 4 mitosis por 10 campos: 2 puntos); y porcentaje de necrosis tumoral (≥10 %: 1
punto). Según esta puntuación, el riesgo se clasifica como bajo (0-3 puntos), intermedio
(4-5 puntos) o alto (6-7 puntos) [9,10]. La resección quirúrgica completa es el pilar fundamental en el tratamiento del
tumor fibroso solitario. Sin embargo, el manejo posoperatorio debe adaptarse individualmente,
considerando los márgenes quirúrgicos y la puntuación de estratificación de riesgo.
Aunque existen datos limitados, en casos avanzados se puede considerar el uso de quimioterapia
y radioterapia neoadyuvante. En el caso de riesgo intermedio a alto con márgenes positivos
e irresecable/inoperable, la RT adyuvante es una opción razonable [11]. Asimismo, el uso de un inhibidor de la tirosina quinasa (TKI) pazopanib podría
considerarse como tratamiento sistémico de primera línea en estos casos [7]. Sin duda, toda decisión de tratamiento debe ser tomada por un equipo multidisciplinario.
4. Conclusión
El presente reporte pone de manifiesto que el diagnóstico temprano, la resección completa
y un plan de seguimiento estructurado son determinantes para lograr resultados clínicos
favorables en los TFS de la pleura. Aunque el tumor presentó un tamaño considerable,
la evolución posoperatoria transcurrió sin complicaciones y el control a largo plazo
no evidenció recurrencia. Estos hallazgos subrayan el papel clave del abordaje multidisciplinario
y la monitorización estrecha en el manejo de esta entidad.