Pharmacological treatment of neoplasms associated with Von Hippel-Lindau disease.
A literature review
Article / Artículo
https://doi.org/10.33821/765
Date received: 04/8/2024
Date accepted: 02/11/2024
Date: 30/12/2024
1. Introduction
Von Hippel-Lindau disease (VHL) is an autosomal dominant syndrome caused by mutations
in the VHL gene. This a tumor suppressor gene located on chromosome 3p25-26; it was
sequenced in 1988 and cloned in 1993, and it encodes the VHL protein (pVHL) [1,2]. About 20% of patients have no family history and present de novo mutations [3]. The incidence is 1/35,000 to 1/45,500 people [4,5]. The clinical manifestations of the disease are hemangioblastomas of the central
nervous system (CNS), 60-80% [6]; retinal hemangioblastomas, 49-62% [7]; endolymphatic sac tumors, 6-15% [8,9]; renal cell carcinoma or renal cysts, 30-70% [7]; Pheochromocytomas (PCC), 10-20% [7]; pancreatic neuroendocrine tumors (pNET) or pancreatic cysts, 35-70% [8]; and epididymal cystadenomas, 25-60% [7].
In 1894, Treacher Collins, a British ophthalmologist, was the first to observe several
lesions in the form of a plexus of blood vessels in the retina of two brothers. He
concluded that it was a new, hereditary disease and called it capillary nevus [10]. Later in 1904, Von Hippel reported retinal events in two patients, with progression
to multiple lesions in one of them, and named the disease angiomatosis retinae [11,12]. In 1926, the pathologist Arvid Lindau described in his monograph the retinal, cerebral,
and visceral components of this disease; to do so, he compiled information from 40
cases, which Cushing called Lindau disease [10,13].
The diagnosis is made when the patient is a carrier of the genetic variant and has
one or more clinical manifestations of the disease, at least one clinical manifestation,
a first-degree relative who is a carrier of the mutation; genetic confirmation is
suggested in patients without a family history, who have a minimum of two types of
tumors associated with the disease, one of them being a hemangioblastoma (HB) [14] .
Although most tumors appear in adulthood, some can develop earlier, e.g., retinal
angioma, pheochromocytoma, and renal cell carcinoma. Therefore, annual clinical/neurological
and retinal surveillance is recommended from birth and for life and imaging studies
of the abdomen and CNS every 2 years, from age 15, with an initial magnetic resonance
imaging (MRI) of the CNS at 10 years of age [14,15]. It is estimated that VHL disease is present in one third of patients with CNS hemangioblastoma,
in more than 50% of retinal angiomas, in 1% of renal cell cancer (RCC), in 50% of
familial pheochromocytoma, and in 11% of apparently sporadic pheochromocytoma [3,16,17].
Renal cell carcinoma occurs in up to 70% of patients with VHL disease. In these cases,
nephron-sparing surgery is recommended when the tumor diameter is ≥3.0 cm due to the
risk of metastasis, and when accelerated tumor growth is evident [18,19]. It results in a recurrence-free survival rate of 76% at 5 years and 20% at 8 years
[20]. In a retrospective study, 181 patients with renal tumors associated with VHL disease
were divided into two groups: Group 1 (108 patients) presented tumors smaller than
3 cm, the mean follow-up was 58 months, and surgery was recommended when the tumor
reached 3 cm, in this group, no metastatic disease was developed; Group 2 (73 patients)
presented a tumor diameter greater than 3 cm, the mean follow-up was 72.9 months and
metastasis occurred in 27.4% of patients. It was concluded that the larger the tumor
size, the greater the risk of metastasis [18].
Regarding tumor growth rate, 41 patients with clear cell renal cell carcinoma (ccRCC)
were evaluated retrospectively. Tumor growth kinetics ranged from 0.24-2.74 cm/year,
with a mean of 0.287 cm/year, patients showed great variation in growth rates: 27.5%
had slow-growing tumors, 44.1% moderate-growing tumors, and 28.4% fast-growing tumors
[21] .
For many years, neoplasms associated with VHL disease were treated by surgical resection
or ablation with the aim of reducing the risk of metastatic disease and controlling
local or systemic sequelae [20,22,27]. An effective systemic alternative could reduce the surgical burden and represents
a new approach to oncologic treatment.
2. Genetics and molecular biology of VHL disease
The VHL protein plays a key role in cellular oxygen sensing. Under normoxic conditions,
it targets hypoxia-inducible factor alpha (HIF- α) for proteasomal degradation. In
hypoxia, HIF- α accumulates, and leads to overproduction of vascular endothelial growth
factor (VEGF), transforming growth factor beta (TGFß), platelet-derived growth factor
(PDGF), glucose transporter 1 (GLUT-1) and erythropoietin (EPO), and promoting angiogenesis,
differentiation, migration and cell proliferation. The HIF-a pathway presents two
isoforms: HIF-1 α and HIF-2α that play distinct roles in response to hypoxia [28,29].
In VHL disease, one allele of the VHL gene is mutated in the germline and the second
allele may be lost somatically (usually through loss of chromosome 3p). Subsequent
loss of pVHL causes HIF- α to accumulate in the absence of hypoxia (referred to as
"pseudohypoxia"), thus resulting in activation of downstream HIF targets and tumorigenesis
of affected tissues [30].
Previous investigation showed that binding small molecules to an internal pocket in
HIF-2 α could allosterically inhibit the protein-protein interaction between HIF-2
α and transcriptional ribonucleic acid (TRNA), which would lead to the inhibition
of transcriptional activity by stopping the processes of tumorigenesis [31-33].
3. Materials and methods 3.1 Main goal
This study aims at establishing the level of efficacy of the different study drugs
in neoplasms associated with Von Hippel-Lindau disease.
3.2 Secundary goal
Determine the type of drug toxicity that led to discontinuation of treatment, and
describe which tumors were most sensitive to the study drugs.
3.3 Study Design
We conducted a descriptive, observational systematic review with a qualitative approach.
3.4 Databases and Terminology Search
We conducted an electronic search for systemic treatment in neoplasms associated with
von Hippel-Lindau disease until July 31, 2024. The search was made in Cochrane Central
Register of Controlled Trials (CENTRAL), PUBMED, and SCIELO databases using MeSH terms
and free text, in addition to the Boolean operators AND, OR and NOT. In PUBMED the
search equation was (((clinical trial) OR (pilot study)) OR (drug therapy)) AND ("von Hippel-Lindau" [Title]), 148 medical articles were found, out of which 5 met the selection criteria to be
part of the study. In COCHRANE, the search equation was "von Hippel Lindau Disease":ti,ab,kw AND Therapy, 14 articles were found and 4 were selected. In addition, a search was performed in
the SCIELO database using the equation ((ab:("von hippel Lindau"))) AND (therapy), 3 articles were found, but none met the selection criteria. A search was attempted
in SCOPUS, but medical articles were not open access.
3.5 Inclusion Criteria
-
- Clinical studies in which the patient sample presented a diagnosis of Von Hippel-Lindau
disease with genetic confirmation, or with clinical characteristics of the disease
but with a family history.
-
- Human clinical trials.
-
- Study participants over 18 years of age.
-
- Studies published by July 31, 2024.
-
- Open access articles.
3.6 Exclusion Criteria
-
- Research reviews, single case studies, books or manuals.
-
- Studies in pediatric patients.
-
- Animal studies.
-
- Studies with symptomatic brain metastases.
-
- Clinical trials with solid tumors not associated with VHL disease.
-
- Research whose patients received surgical treatments, radiotherapy, radiosurgery
or radiofrequency ablation.
-
- Studies aimed solely at the treatment of hemangioblastomas, or retinal hemangiomas
associated with VHL disease.
3.7 Selection of articles
We selected clinical studies evaluating the effectiveness and toxicity of a drug in
a sample of patients with tumors associated with Von Hippel Lindau disease. The information
was selected by title and abstract in electronic databases. Then, full-text articles
were downloaded, read, and those that included clinical trials with systemic treatment
for neoplasias associated with VHL disease were selected. Data were extracted in a
table and the methodological quality was analyzed. The heterogeneity of these studies
was assessed. There were no restrictions on language or publication status (Figura 1).
Figure 1
PRISMA 2020 Flowchart
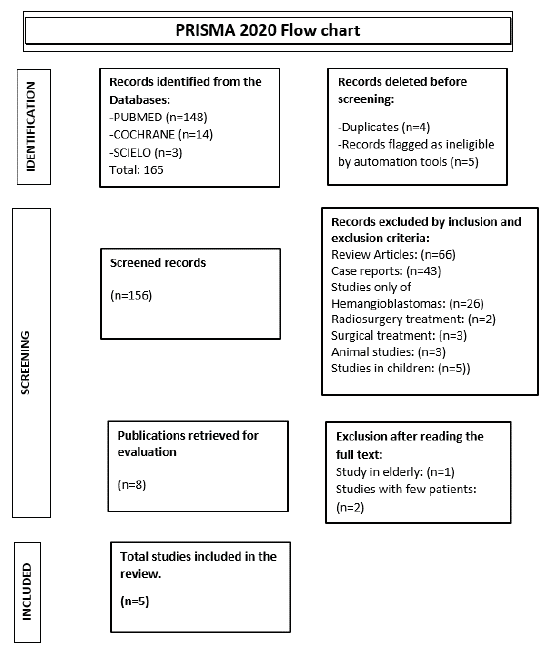
Source: Own elaboration based on PRISMA 2020 flow chart
4. Results and Discussion
Table 1 shows the characteristics of the four prospective studies and the retrospective article,
with a small sample in each study.
Currently, the majority of patients with neoplasias associated with VHL disease undergo
repetitive surgical procedures, leading to neurological sequelae, renal or pancreatic
failure, and a decline in their quality of life. As a result, targeted therapy studies
have been done on VGFR, FGFR, and HIF-2 α, and the results have been positive in terms
of how well it works and how safe it is.
Table 1
Characteristics of the studies
|
Author
|
Author Study design
|
Number of patients. Manifestations of VHL disease
|
Assessment criteria
|
Median age in years
|
Methodology (Intervention - dose)
|
Median follow-up
|
Responses according to RECIST
|
Median time to RECIST response
|
Adverse events
|
Observations
|
|
Jonasch E et al., 2011 [34]
|
Prospective, open-label, single-arm phase II study
|
N = 15 6 discontinued treatment 12: CCR. 11: Renal cysts. 11: CNS HB. 9: Retinal hemangiomas.
7: Pancreatic cysts. 7: Neuroendocrine tumors of the pancreas. 3: Adrenal lesions.
2:Endolymphatic sac tumor. 1: Cystadenoma of epididymis. N = 14 No. of injuries in
the study 11: Hemangioblastomas of the cerebellum. 11: Pancreatic cysts. 9: Renal
cysts. 8: Spinal
|
Main objective: security. Secondary objective: Efficacy of complete responses (CR)
+ partial responses (PR).
|
36 (22-57)
|
Sunitinib 50 mg/ day for 28 days, followed by 14 days of rest , for 4 cycles, with
the possibility of dose reduction to 37.5 or 25 mg due to toxicity.
|
48 weeks
|
33% of RCC responded partially to none of the HBs ( P = 0.014) . HB: 91% stable disease.
The rest of the injuries were stable.
|
|
Fatigue, handfoot syndrome, nausea, grade 3 neutropenia.
|
At 48 weeks, RCC and neuroendocrine tumors had grown again, reaching measurements
close to the initial ones, but not larger.
|
|
Roma A et al., 2015 [35]
|
Retrospective Study
|
hemangioblastomas 7: Retinal angiomas. 3: Neuroendocrine tumors of the pancreas. 3:
Epididymal cysts. 2: Pheochromocytoma. 1: Supratentorial hemangioblastoma. 1: Adrenal
adenoma
|
Secondary objectives: radiological response, toxicity and overall survival (OS)
|
48 (27-71)
|
Sunitinib 50 mg/ day for 28 days, followed by 14 days of rest , with the possibility
of dose reduction to 37.5 or 25 mg due to toxicity.
|
39.4 months
|
RP: 64.3% of patients. Stable disease: 35.7% SLP: 85.7% the first year. 71.4% in the
second year. With the exception of HB, there was a response in all lesions.
|
|
Mucositis, handfoot syndrome, asthenia, hypertension, hypothyroidism.
|
Patients received a median of 19.5 cycles, treatment was continued until maximum response,
progression, unacceptable toxicity, or patient refusal.
|
|
Pilié P et al., 2018 [36]
|
Prospective, single-center, open-label, single-arm, phase 2 study.
|
N = 6 3 discontinued treatment 5: HB in cerebellum. 4: HB of the brain stem. 3: Retinal
HB 2: RCC 2: Pancreatic cysts.
|
Main objective: security. Secondary objectives: Effectiveness
|
44 (18-61)
|
Dovitinib 500 mg/ day in a 4-week cycle with a 5-day on, 2-day off schedule, for 6
cycles.
|
|
No RECIST response was evident. The HBs showed stability.
|
|
Rash, diarrhea, fatigue.
|
The study was discontinued due to toxicity.
|
|
Jonasch E et al., 2018 [37]
|
Prospective, single-arm, phase 2 study.
|
23: CNS lesions. 22: Kidney injuries. 9: Lesions in the pancreas. 3: Eye injuries.
1:Lesions in Adrenal.
|
Objective response rate (ORR) and safety.
|
38 (32- 42).
|
Pazopanib 800 mg/day, with dose reduction in 200 mg increments permitted if patients
experienced grade 3 or higher toxicity for 24 weeks.
|
12 months
|
RP: 42 %. Stable disease: 58%. TRO by organ: RCC: 52%. Pancreatic lesions: 53%. CNS HB: 4%.
|
RCC: 3 months. HB: 6 months. Pancreatic lesions: 6 months
|
Fatigue, diarrhea or transaminitis.
|
Most patients decided to continue treatment after 24 weeks.
|
|
Jonasch E et al., 2021 [38]
|
Prospective, open-label, single-arm phase 2 study.
|
N = 61 7 discontinued treatment. All patients had RCC and pancreatic lesions. 22:Neuroendocrine
tumors of the pancreas. 50: CNS HB. 12: Retinal HB: 12.
|
Main objective: objective response. Secondary objectives: duration of response, time
to response and progressionfree survival in RCC, other criteria were efficacy in non-renal
carcinomas associated with VHL disease and safety of Belzutifan.
|
41 (19-66),
|
Belzutifan 120 mg/day.
|
21.8 months
|
TRO by organ: RCC: 49%. Neuroendocrine tumors of the pancreas: 91%. CNS HB: 30% The
median duration of response was not reached.
|
RCC: 8.2 months. Pancreatic neuroendocrine tumors: 5.5 months. CNS HB: 3.2 months.
|
Anemia grade 1 and 2 (90%), Fatigue grade 1 and 2 (66%).
|
54 Patients (89%) were still receiving treatment with Belzutifan at the data cut-off
date. Prior to the initiation of Belzutifan, patients had undergone 327 procedures
(surgery, radiofrequency ablation), out of which 64 occurred in the 2.5 years prior
to the start of the study, and only three surgeries were required during the 22 months
of Belzutifan.
|
The study conducted by Jonasch et al. in 2011 [34] revealed that the growth of RCC and pancreatic NETs recurred after 48 weeks, a maximum
of four treatment cycles, despite prolonged administration of sunitinib, as reported
by Rome. According to the response evaluation criteria for solid tumors (RECIST),
most patients in a 2015 study maintained their responses, suggesting the need for
additional research to determine the best doses for prolonged treatments, given the
potential for adverse events that could result in treatment abandonment [34, 35].
The analysis of samples from the Tissue Bank of MD Anderson at the University of Texas
revealed a null response of the HB to sunitinib, a VEGF receptor inhibitor, with low
expression of the VEGF receptor 2 in the HB compared to the RCC (p = 0.003), and higher
levels of the fibroblast growth factor receptor substrate 2 (FGF) in the HB (p = 0.003).
This raises the hypothesis that treatment with FGF receptor blockers may benefit patients
with HB [34].
It is unclear why VHL-derived malignancies respond differently to therapy. It looks
like sunitinib is a better way to treat VHL-related RCC than other VHL-related lesions.
However, its side effects have made it hard to use for long periods of time [34, 35].
By changing the molecular mechanism that affects clinical manifestations, the ultimate
goal of systemic therapy is to lower the number of surgeries needed in people with
VHL disease [30,32,33].
The review's analysis reveals that HIF-2 inhibition provides a superior safety and
efficacy profile compared to the antiangiogenic agents sunitinib and pazopanib. On
August 13, 2021, the Food and Drug Administration (FDA) approved belzutifan for patients
with VHL who have developed clear-cell renal cell carcinoma, central nervous system
HB, or pancreatic neuroendocrine tumors associated with the disease [39].
The belzutifan clinical trial results suggest that it could be used instead of or
in addition to surgery to treat people with VHL disease. This is because it delays
or eliminates the need for surgeries that are linked to serious problems like neurological
sequelae or renal or pancreatic failure. It also lowers surgical morbidity and breaks
the cycle of having to have surgeries over and over again [38, 40].
Reporting response in individual lesions is not a common way to share the results
of a clinical study. However, for people with VHL disease, each clinical manifestation
is a separate medical and surgical challenge, and a drug effect on any lesion may
mean that surgery is not necessary. Limitations of the review for extrapolating the
results to the general population include the limited number of studies, the heterogeneity
of the samples, and the low number of patients per study. However, we should acknowledge
that there remains a vast field to delve into. The studies that were looked at suggest
that histopathological samples from people with VHL disease should be studied to find
out the molecular differences in the affected tissues and then target therapies should
be looked into.
Limitations of the review to extrapolate the results to the general population include
the low number of studies, heterogeneity of the samples, and low number of patients
per study. However, there is still a wide field to explore; the analyzed studies suggest
the investigation of histopathological samples associated with VHL disease to determine
the molecular differences of the affected tissues and subsequently investigate target
therapies.
5. Conclusions
Mutations in patients with Von Hippel-Lindau disease (VHL) demand continuous surveillance
and a multidisciplinary therapeutic approach. Repeated surgical interventions cause
serious physical and psychological sequelae; thus, systemic management seeks to prevent
invasive procedures and improve quality of life, especially in cases of associated
neoplasia.
HIF-2a inhibition has shown a better safety and efficacy profile compared to traditional
antiangiogenic agents, thus minimizing serious adverse events. Since its FDA approval
in 2021, Belzutifan has revolutionized the treatment of renal carcinoma and other
VHL-associated neoplasms by stopping or reversing tumor growth, reducing the need
for surgery and the risk of metastasis.
1. Introducción
La enfermedad de von Hippel-Lindau (VHL) es un síndrome autosómico dominante causado
por mutaciones en el gen de VHL, un gen supresor de tumores que se encuentra en el
cromosoma 3p25-26. Fue secuenciado en 1988 y clonado en 1993, y codifica la proteína
VHL (pVHL) [1,2]. Alrededor del 20 % de los pacientes no tienen antecedentes familiares y presentan
una mutación de novo [3]. La incidencia es de 1/35 000 a 1/45 500 personas [4,5]. Las manifestaciones clínicas de la enfermedad son hemangioblastomas (HB) del sistema
nervioso central (SNC): 60-80 % [6]; HB de retina: 49-62 % [7]; tumores del saco endolinfático: 6-15 % [8,9]; carcinoma de células renales o quistes renales: 30-70 % [7]; feocromocitomas (PCC): 10-20 % [7]; tumores neuroendocrinos pancreáticos (pNET) o quistes pancreáticos: 35-70 % [8]; y cistadenomas epididimarios: 25-60 % [7].
En 1894, Treacher Collins, un oftalmólogo británico, fue el primero en observar varias
lesiones en forma de plexo de vasos sanguíneos en la retina de dos hermanos y concluyó
que se trataba de una enfermedad nueva, hereditaria, y la denominó nevus capilar [10]. Posteriormente, en 1904, von Hippel informó eventos retinianos en dos pacientes,
con progresión a múltiples lesiones en uno de ellos y bautizó la enfermedad como angiomatosis
retinae [ 11,12]. En 1926, el patólogo Arvid Lindau describió en su monografía los componentes retiniano,
cerebral y visceral de esta enfermedad, para lo cual recopiló la información de 40
casos, que Harvey Cushing denominó enfermedad de Lindau [10,13].
El diagnóstico se realiza cuando el paciente es portador de la variante genética y
tiene una o más manifestaciones clínicas de la enfermedad; en personas con al menos
una manifestación clínica y que tengan un familiar en primer grado portador de la
mutación; además, se sugiere confirmación genética en pacientes sin antecedentes familiares
y que tengan mínimo dos tipos de tumores asociados a la enfermedad, en los que uno
sea un HB [14].
A pesar de que la mayoría de los tumores se manifiestan en la edad adulta, existen
algunos que pueden desarrollarse antes, como el angioma retiniano, el feocromocitoma
y el carcinoma de células renales. Por lo tanto, se recomienda vigilancia clínica/neurológica
y de retina anual desde el nacimiento y de por vida, y estudios de imagen del abdomen
y del SNC cada dos años, desde los 15 años, con una resonancia magnética (RM) inicial
del SNC a los 10 años de edad [14,15]. Se estima que la enfermedad de VHL se encuentra en un tercio de los pacientes con
HB del SNC en más del 50 % de los angiomas de retina; en el 1 % de cáncer de células
renales (RCC); en el 50 % de los feocromocitomas familiares y en el 11 % de los feocromocitomas
aparentemente esporádicos [3,16,17].
El carcinoma de células renales se presenta hasta en el 70 % de los pacientes con
enfermedad de VHL; en estos casos se recomienda la cirugía conservadora de nefronas
cuando el diámetro del tumor es >3,0 cm, por el riesgo de metástasis, y cuando se
evidencia un crecimiento tumoral acelerado [18,19]. Esto último resulta en una tasa de supervivencia libre de recurrencia del 76 %
a los 5 años y del 20 % a los 8 años [20]. En un estudio retrospectivo, 181 pacientes con tumores renales asociados a la enfermedad
de VHL fueron divididos en dos grupos. El grupo 1 (108 pacientes) presentó tumores
de tamaño menor a 3 cm, la media de seguimiento fue de 58 meses y cuando el tumor
alcanzó los 3 cm se recomendó la cirugía; en este grupo no se desarrolló enfermedad
metastásica. El grupo 2 (73 pacientes) presentó un diámetro tumoral mayor a 3 cm.
La media de seguimiento fue de 72.9 meses y se presentó metástasis en el 27,4 % de
los pacientes. Finalmente, se concluyó que a mayor tamaño tumoral mayor riesgo de
metástasis (18).
En cuanto a la velocidad de crecimiento tumoral, se evaluaron retrospectivamente 41
pacientes con carcinoma renal de células claras (ccRCC), la cinética de crecimiento
tumoral osciló entre 0,242,74 cm/año, con una media de 0,287 cm/año; los pacientes
presentaron gran variación en las tasas de crecimiento: el 27,5 % tuvo tumores de
crecimiento lento; el 44,1%, tumores de crecimiento moderado y el 28,4 %, tumores
de crecimiento rápido [21].
Durante muchos años, las neoplasias asociadas a la enfermedad de VHL fueron tratadas
mediante resección quirúrgica o ablación con el objetivo de reducir el riesgo de enfermedad
metastásica y controlar las secuelas locales o sistémicas [20,22-27]. Una alternativa sistémica eficaz podría reducir la carga quirúrgica y representa
un nuevo enfoque para el tratamiento oncológico.
2. Genética y biología molecular de la enfermedad de VHL
La proteína VHL desempeña un papel clave en la detección del oxígeno celular, en condiciones
de normoxia se dirige al factor inducible por hipoxia alfa (HIF-α) para su degradación proteasomal. En evidencia de hipoxia, el HIF- α se acumula, y esto conduce a la sobreproducción del factor de crecimiento del endotelio
vascular (VEGF), del factor de crecimiento transformador beta (TGFß), del factor de
crecimiento derivado de plaquetas (PDGF), del transportador de glucosa 1 (GLUT-1)
y de la eritropoyetina (EPO), lo que promueve la angiogénesis, la diferenciación,
la migración y la proliferación celular. La vía HIF- α presenta dos isoformas: la HIF-1 α y la HIF-2α, que desempeñan papeles distintos
en respuesta a la hipoxia [28,29].
En la enfermedad de VHL, un alelo del gen de VHL está mutado en la línea germinal
y el segundo alelo puede perderse somáticamente, por lo general, a través de la pérdida
del cromosoma 3p. La pérdida posterior de la pVHL hace que el HIF- α se acumule en ausencia de hipoxia (lo que se conoce como "pseudohipoxia"), y que
resulte en la activación de los objetivos del HIF en sentido descendente y la tumorogénesis
de los tejidos afectados [30].
Durante una investigación se demostró que la unión de moléculas pequeñas a un bolsillo
interno en HIF-2 α podía inhibir alostéricamente la interacción proteína-proteína
entre el HIF-2 α y el ácido ribonucleico transcripcional (ARNT), lo que llevaría a
la inhibición de la actividad transcripcional, y detendría los procesos de tumorogénensis
[31-33].
3. Materiales y métodos
3.1 Objetivo primario
Establecer el nivel de eficacia de los diferentes fármacos del estudio en las neoplasias
asociadas con la enfermedad de VHL.
3.2 Objetivos secundarios
Determinar el tipo de toxicidad farmacológica que llevó al abandono del tratamiento
y describir cuáles fueron los tumores más sensibles a los fármacos del estudio.
3.3 Diseño del estudio
Se realizó una revisión sistemática descriptiva, observacional, con un enfoque cualitativo.
3.4 Bases de datos y terminología de la búsqueda
Se efectuó una búsqueda electrónica en las bases de datos del Registro Cochrane Central
de Ensayos Controlados (CENTRAL Central), en PUBMED y en SciELO. La búsqueda electrónica,
efectuada hasta el 31 de julio del 2024 acerca del tratamiento sistémico en las neoplasias
asociadas con la enfermedad de VHL, se efectuó en tres bases de datos y se limitó
con los operadores booleanos, términos MESCH y texto libre. En PubMed la ecuación
de búsqueda fue (((clinical trial) OR (pilot study)) OR (drug therapy)) AND ("von
Hippel-Lindau" [Title]); se encontraron 148 artículos médicos, de los cuales cinco
cumplieron los criterios de selección para formar parte del estudio. En CENTRAL la
ecuación de búsqueda fue "von Hippel Lindau Disease":ti,ab,kw AND Therapy; se encontraron
14 artículos y se seleccionaron cuatro. En la base de datos SciELO, se utilizó la
ecuación ((ab:("von hippel Lindau"))) AND (therapy) y se encontraron tres artículos,
pero ninguno cumplió los criterios de selección para ser parte del estudio. Se intentó
realizar una búsqueda en Scopus, pero no se tuvo acceso libre a los artículos médicos.
3.5 Criterios de inclusión
Se incluyeron estudios clínicos cuya muestra de pacientes presentó el diagnóstico
de enfermedad de VHL, con confirmación genética o con características clínicas de
la enfermedad, pero con antecedentes familiares. Ensayos clínicos en humanos. Los
participantes de los estudios debían tener más de 18 años. Estudios que se hubieran
publicado hasta el 31 de julio del 2024. Artículos con acceso a texto completo.
3.6 Criterios de exclusión
Se excluyeron revisiones, estudios de caso único, libros o manuales. Estudios en pacientes
pediátricos. Estudios en animales. Estudios con metástasis cerebrales sintomáticas.
Ensayos clínicos con tumores sólidos no asociados con la enfermedad de VHL. Investigaciones
cuyos pacientes recibieron tratamientos quirúrgicos, radioterapia, radiocirugía o
ablación por radiofrecuencia. Aquellos estudios dirigidos únicamente al tratamiento
de HB o hemangiomas de retina asociados a la enfermedad de VHL.
3.7 Selección de artículos
Se escogieron los estudios clínicos en los que se evaluó la efectividad y la toxicidad
de un medicamento en una muestra de pacientes con tumores asociados con la enfermedad
de VHL. La información se seleccionó por título y resumen en bases de datos electrónicas;
después, los artículos se descargaron a texto completo, se leyeron y, por último,
se escogieron todos los que incluían ensayos clínicos con tratamiento sistémico para
las neoplasias asociadas con la enfermedad de VHL. Los datos extraídos se depositaron
en una tabla y se analizó la calidad metodológica. Se valoró la heterogeneidad de
esos estudios. No hubo restricciones por idioma o estado de publicación. Luego de
su lectura en profundidad, se seleccionaron cinco estudios para la revisión, de los
cuales cuatro son estudios prospectivos y uno es retrospectivo (Figura 1).
Figura 1
Diagrama de flujo PRISMA 2020
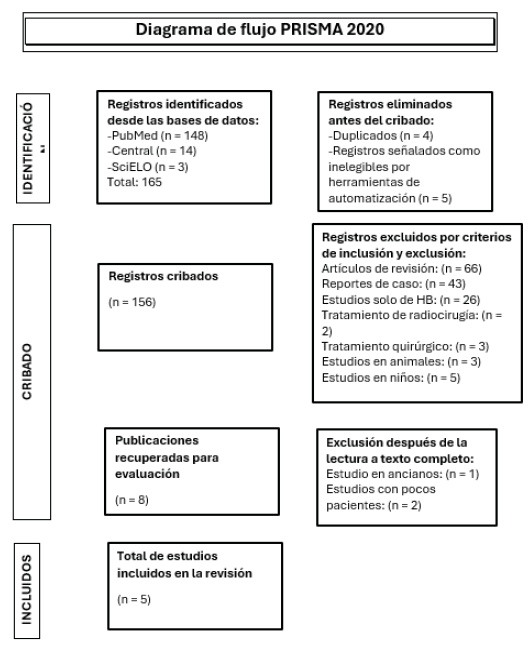
Fuente: Elaboración propia con base en el flujograma PRISMA 2020
4. Resultados y discusión
En la tabla 1 se muestran las características de los cuatro estudios prospectivos y el artículo
retrospectivo, con una muestra pequeña en cada estudio.
Actualmente, la mayoría de pacientes con neoplasias asociadas con la enfermedad de
VHL se somete a procedimientos quirúrgicos repetitivos, lo que ha conllevado secuelas
neurológicas e insuficiencia renal o pancreática, con deterioro de la calidad de vida
de los pacientes. Frente a esto, se han realizado estudios clínicos con terapia target,
dirigida al VGFR, FGFR y al HIF-2α, que han arrojado resultados alentadores de eficacia
y seguridad.
Tabla 1
Características de los estudios
|
Autor
|
Diseño del estudio
|
N.° de pacientes. Manifestaciones de la enfermedad de VHL
|
Criterios de valoración
|
Mediana de edad en años.
|
Metodología (intervencióndosis)
|
Mediana de seguimiento
|
Respuestas según RECIST
|
Mediana de tiempo hasta la respuesta RECIST
|
Eventos adversos
|
Observacione
|
|
Jonasch E et al., 2011 [34]
|
Estudio prospectivo de fase II, abierto y de un solo brazo.
|
N = 15 6 interrumpieron el tratamiento 12: CCR. 11: Quistes renales. 11: HB del SNC.
9: Hemangiomas de retina. 7: Quistes pancreáticos. 7: Tumores neuroendocrinos del
páncreas. 3: Lesiones suprarrenales. 2:Tumor del saco endolinfático. 1: Cistoadenoma
del epidídimo. N = 14 n.° de lesiones en el estudio
|
Objetivo principal: seguridad. Objetivo secundario: eficacia respuestas completas
(RC) + respuestas parciales (RP).
|
36 (22-57)
|
Sunitinib 50mg/ día durante 28 días, seguidos de 14 días de descanso, por 4 ciclos,
con posibilidad de reducción de la dosis a 37,5 o 25 mg por toxicidad.
|
48 semanas
|
El 33 % de RCC respondieron parcialmente frente a ninguno de los HB (p = 0,014). HB:
91 % enfermedad estable. Las demás lesiones presentaron estabilidad.
|
|
Fatiga, síndrome mano-pie, náusea, neutropenia grado 3.
|
A las 48 semanas el RCC y los tumores neuroendocrinos habían vuelto a crecer, hasta
alcanzar medidas cercanas a las iniciales, pero no mayores.
|
|
Roma A et al., 2015 [35]
|
Estudio retrospectivo.
|
11: HB del cerebelo. 11: Quistes pancreáticos. 9: Quistes renales. 8: HB espinales.
7: Angiomas de retina. 3: Tumores neuroendocrinos del páncreas. 3: Quistes del epidídimo.
2: Feocromocitoma. 1: HB supratentorial. 1: Adenoma adrenal.
|
Objetivo principal: supervivencia libre de progresión (SLP). Objetivos secundarios:
respuesta radiológica, toxicidad y supervivencia global (SG).
|
48 (27-71)
|
Sunitinib 50mg/ día durante 28 días, seguidos de 14 días de descanso, con posibilidad
de reducción de la dosis a 37,5 o 25 mg por toxicidad.
|
39.4 meses
|
RP: 64,3 % de pacientes. Enfermedad estable: 35,7 % SLP: 85,7 % el primer año. 71,4
% el segundo año. Con excepción de los HB, hubo respuesta en todas las lesiones.
|
|
Mucositis, síndrome de mano pie, astenia, hipertensión, hipotiroidismo.
|
Los pacientes recibieron una media de 19,5 ciclos, el tratamiento fue continuado hasta
la respuesta máxima, progresión, toxicidad inaceptable, o rechazo del paciente.
|
|
Pilié P et al., 2018 [36]
|
Estudio prospectivo de fase 2, de un solo centro, abierto y de un solo brazo.
|
N = 6 3 interrumpieron el tratamiento 5: HB en cerebelo. 4: HB del tronco encefálico.
3: HB retinianos. 2: RCC. 2: Quistes pancreáticos. N = 31 7 interrumpieron el tratamiento
|
Objetivo principal: seguridad. Objetivos secundario: eficacia.
|
44 (18-61)
|
Dovitinib 500 mg/ día en un ciclo de 4 semanas con un esquema de 5 días de tratamiento
y 2 días de descanso, por 6 ciclos.
|
|
No se evidenció respuesta RECIST. Los HB presentaron estabilidad.
|
|
Sarpullido, diarrea, fatiga
|
El estudio se suspendió por toxicidad.
|
|
Jonasch E et al., 2018 [37]
|
Estudio prospectivo de fase 2, de un solo brazo.
|
23: Lesiones en SNC. 22: Lesiones en riñón. 9: Lesiones en páncreas. 3: Lesiones en
ojos. 1:Lesiones en suprarrenal.
|
Tasa de respuesta objetiva (TRO) y seguridad.
|
38 (32- 42).
|
Pazopanib 800 mg/día, con una reducción de la dosis en incrementos de 200 mg permitida
si los pacientes presentaban toxicidad de grado 3 o mayor. Esto durante 24 semanas
|
12 meses
|
RP: 42 %. Enfermedad estable: 58 %. TRO por órgano: RCC: 52 %. Lesiones pancreáticas:
53 %. HB del SNC: 4 %.
|
RCC: 3 meses. HB: 6 meses. Lesiones pancreáticas: 6 meses
|
Fatiga, diarrea o transaminitis.
|
La mayoría de los pacientes decidieron continuar con el tratamiento después de 24
semanas.
|
|
Jonasch E et al., 2021 [38]
|
Estudio prospectivo de fase 2, abierto y de un solo brazo.
|
N = 61 7 interrumpieron el tratamiento. Todos los pacientes tenían RCC y lesiones
pancreáticas. 22:Tumores neuroendocrinos del páncreas. 50: HB del SNC. 12: HB de retina:
12
|
Objetivo principal: respuesta objetiva. Objetivos secundarios: duración de la respuesta,
el tiempo hasta la respuesta y la supervivencia libre de progresión en RCC, otros
criterios fueron la eficacia en carcinomas no renales asociados con la enfermedad
de VHL y la seguridad del belzutifan.
|
41 (19-66),
|
Belzutifan 120 mg/día.
|
21.8 meses
|
TRO por órgano: RCC: 49 %. Tumores neuroendocrinos del páncreas: 91 %. HB del SNC:
30 % La mediana de duración de respuesta no fue alcanzada
|
RCC: 8.2 meses. Tumores neuroendocrinos pancreáticos: 5.5 meses. HB del SNC: 3.2 meses
|
Anemia grado 1 y 2 (90 %), Fatiga grado 1 y 2 (66 %)
|
54 Pacientes (89 %) seguían recibiendo tratamiento con belzutifan a la fecha de corte
de los datos. Antes del inicio del belzutifan, los pacientes se habían realizado 327
procedimientos (cirugía, ablación por radiofrecuencia), de los cuales 64 en los 2,5
años anteriores al inicio del estudio, y solo fueron necesarias tres cirugías durante
los 22 meses del belzutifan.
|
Los resultados del estudio de Jonasch et al., 2011 [34] muestran que los RCC y los NET pancreáticos habían vuelto a crecer luego de 48 semanas,
después de máximo cuatro ciclos de tratamiento; mientras que la administración prolongada
de sunitinib, según el estudio de Roma A, publicado en el años 2015, mantuvo respuestas,
según los criterios de evaluación de respuesta en tumores sólidos (RECIST), prolongadas
en la mayoría de pacientes, lo que sugirió nuevos estudios en el hallazgo de dosis
óptimas para tratamientos prolongados, debido a los eventos adversos que pueden producirse
y que llevan al abandono del tratamiento [34,35].
En los estudios con sunitinib, un inhibidor del receptor del VEGF, se encontró una
nula respuesta de los HB a este medicamento, lo que tendría relación con el análisis
de muestras del Banco de Tejidos del MD Anderson de la Universidad de Texas, donde
se observó una baja expresión del receptor 2 del VEGF en el HB en relación con el
RCC (p = 0,003), mientras que los niveles del sustrato 2 del receptor del factor de
crecimiento de fibroblastos (FGF) fue superior en los HB (p = 0,003). Esto plantea
la hipótesis de que el tratamiento con bloqueadores del receptor del FGF puede beneficiar
a los pacientes con HB [34].
No está claro por qué las neoplasias derivadas de VHL responden de manera diferente
a la terapia. El sunitinib parece ser una opción de tratamiento más apropiada para
el CCR relacionado con VHL que para otras lesiones asociadas con VHL; sin embargo,
su toxicidad ha sido un factor limitante de relevancia para tratamientos prolongados
[34,35].
El objetivo final de la terapia sistémica es disminuir la cantidad de intervenciones
quirúrgicas en pacientes con enfermedad de VHL, manipulando el mecanismo molecular
subyacente que afecta las manifestaciones clínicas [30,32,33].
A partir del análisis de los estudios incluidos en la revisión, se observa que la
inhibición del HIF-2α ofrece un mejor perfil de seguridad y eficacia que los agentes
antiangiogénicos sunitinib y pazopanib. El 13 de agosto del 2021, la Food and Drug
Administration (FDA) aprobó el belzutifan para pacientes con enfermedad de VHL que
hayan desarrollado un carcinoma renal de células claras, HB del sistema nervioso central
o tumores neuroendocrinos del páncreas asociados con la enfermedad [39].
Los resultados del ensayo clínico de belzutifan sugieren que este podría servir como
tratamiento alternativo o un complemento al tratamiento quirúrgico en pacientes con
enfermedad de VHL, pues retrasa o elimina el requerimiento de cirugías que se asocian
con complicaciones importantes como secuelas neurológicas o insuficiencia renal o
pancreática, además de disminuir la morbilidad quirúrgica y romper el ciclo de cirugías
interminables [38,40].
Reportar la respuesta en lesiones individuales no es un mecanismo común para informar
los resultados de un estudio clínico; sin embargo, en individuos con enfermedad de
VHL, cada manifestación clínica es un desafío médico y quirúrgico independiente, y
un efecto farmacológico en cualquier lesión puede reducir la necesidad de intervención
quirúrgica.
Entre las limitaciones de la revisión para extrapolar los resultados a la población
general, se encuentra el número limitado de estudios, la heterogeneidad de las muestras
y el bajo número de pacientes por cada estudio. No obstante, hay que tener en cuenta
que aún queda un amplio campo por explorar. Los estudios analizados sugieren la investigación
de muestras histopatológicas asociadas a la enfermedad de VHL para determinar las
diferencias moleculares de los tejidos afectados y, posteriormente, investigar terapias
target.
5. Conclusiones
El hallazgo de mutaciones en pacientes con enfermedad de VHL demanda vigilancia continua
y un enfoque terapéutico multidisciplinario. Las intervenciones quirúrgicas repetidas
causan secuelas físicas y psicológicas graves. El manejo sistémico busca prevenir
procedimientos invasivos y mejorar la calidad de vida, especialmente en casos de neoplasias
asociadas.
La inhibición de HIF-2α ha mostrado un mejor perfil de seguridad y eficacia en comparación
con los agentes antiangiogénicos tradicionales, lo que minimiza eventos adversos graves.
Desde su aprobación por la FDA en el 2021, el belzutifan ha revolucionado el tratamiento
del carcinoma renal y otras neoplasias asociadas a VHL, pues detiene o revierte el
crecimiento tumoral y reduce la necesidad de cirugía y el riesgo de metástasis.